HEMOGLOBINOPATÍAS
DIAGNÓSTICO DE TALASEMIAS EN NIÑOS
Te has preguntado alguna vez, ¿por qué muchos niños tratados con anemia ferropénica no mejoran con la terapia de hierro?
Con la asesoría de la Dra. Iliana De Los Reyes Valencia y Paula Carolina Guzmán Cruz. Servicio de Hematooncología Pediátrica del Hospital Militar Central (HOMIL).
Tal vez podrías estar frente a un caso de Talasemia, en este blog te enseñamos a identificarla, usted hace parte de este proceso.
¿Sabes qué es la hemoglobina?
La hemoglobina es una proteína compuesta, es decir está constituida por cadenas polipeptídicas de aminoácidos, que constituyen las cadenas de globina y un grupo prostético que no contiene aminoácidos y se llama Hemo, el cual le da la connotación de Hemoproteína (hay un grupo hemo por cada cadena de globina) encargado de darle el color rojo a la hemoglobina, al eritrocito y a la sangre y capturar la molécula de oxígeno.
Las cadenas polipeptídicas son cuatro, razón por la cual se considera a la hemoglobina una proteína tetrámerica, además tiene forma esférica lo que le permite estar dentro de la clasificación de las proteínas globulares.
Las cadenas polipéptidicas de la hemoglobina son Alfa 1 (α1) y Alfa 2 (α2), estas tienen aproximadamente 141 aminoácidos y están ubicadas en el brazo corto del cromosoma 16; las otras cadenas polipéptidicas son las cadenas Beta 1 (β1) y Beta 2 (β2), que tienen 146 aminoácidos y se ubican en el brazo corto del cromosoma 11.
Las cadenas polipeptídicas se pliegan en el 75% de la estructura globular de la hemoglobina en forma de alfa hélices y existen 7 plegamientos alfa hélices para las cadenas Alfas y 8 plegamientos alfa hélices para las cadenas Betas.
Estos plegamientos alfa hélices, son importantes porque mantienen la estructura globular de la globina y sí en ellos ocurre un error en el plegado o hay ausencia de una de las cadenas se pueden presentar enfermedades de las cadenas de la globina, por ejemplo, sí esta alteración se presenta en las cadenas Betas se desencadenan las Betatalasemias y si ocurre en las alfas las Alfatalasemias.
Existen otros defectos puntuales y se llaman mutaciones puntuales, como las que suceden en las cadenas betas donde al cambiar un aminoácido en la posición 6 de esta cadena, ubicada en el cromosoma 11, se modifica la estructura globular de la hemoglobina, porque se hizo un cambio del ácido glutámico por valina y esto origina la Anemia Falciforme o Drepanocitosis.
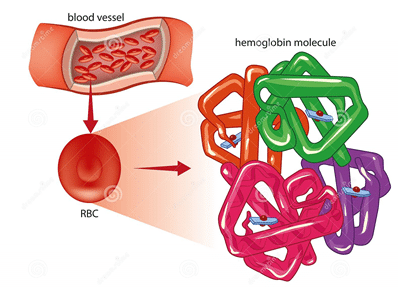
Figura 1.Representación de la hemoglobina
¿Ahora, puedes entender qué son las hemoglobinopatías?
Las hemoglobinopatías son un grupo de enfermedades hereditarias poco comunes que comprometen la estructura normal y función de la molécula de hemoglobina.
Estamos seguros, que ya puedes intuir la respuesta a la siguiente pregunta: ¿Cuántos tipos de hemoglobinopatías existen?
Existen dos tipos de hemoglobinopatías y las vamos a clasificar en:
Variantes estructurales: alteración en la calidad de la hemoglobina, aquí citamos una de las más frecuentes en nuestro medio, la Anemia de Células Falciformes en la que se encuentra presente la hemoglobina S.- Variantes cuantitativas: alteración en la cantidad de las globinas, donde ocurre una reducción en la síntesis y/o desequilibrio en la cantidad de cadenas α y β globinas; a este grupo pertenecen las Talasemias.
¿Puedes comprender ya, qué es la Talasemia?
La Talasemia es un tipo de hemoglobinopatías donde existe una alteración en la cantidad de las cadenas de globina, es decir hay déficit o ausencia de una de las cadenas de globina alfa o beta, lo cual causa que la cadena que no tiene alteración produzca un exceso dentro del glóbulo rojo en desarrollo (porque esta cadena no se aparea), generando precipitación de la hemoglobina y destrucción precoz de los glóbulos rojos antes de alcanzar su maduración completa; todo esto se manifiesta como hemólisis, que no es más que la destrucción prematura del glóbulo rojo.
De esta manera, si las cadenas de α-globina no se producen en cantidades adecuadas, habrá una acumulación de cadenas de β-globina (α-talasemia) y si las cadenas de β-globina se producen inadecuadamente, entonces las cadenas de α-globina se acumularán (β-talasemia).
¿Estás preparado para identificar con base a la severidad clínica, los pacientes talasémicos?
Cuando el glóbulo rojo se destruye antes de alcanzar su vida media normal de 120 días, da como resultado anemia, que es la disponibilidad reducida de la hemoglobina para transportar el oxígeno y la producción de muchos eritrocitos no saludables en la cavidad de la médula ósea donde normalmente se producen; esta condición hace referencia a lo que llamamos producción ineficaz de glóbulos rojos o eritropoyesis ineficaz.
La anemia generalmente resulta en la necesidad de ser corregida mediante transfusión de sangre, es decir hemocomponentes.
En las formas más graves de Betatalasemia, la anemia es tan grave que, a menos que se trate regularmente mediante transfusión de sangre, el paciente morirá temprano en la vida, principalmente en la infancia. La condición se conoce como talasemia dependiente de transfusión o TDT. Otros casos pueden sobrevivir con transfusiones de sangre ocasionales o sin ellas, conocidas como talasemia sin transfusión o NTDT.
Con base a lo anterior, ¿Tienes la información para deducir que la Talasemia es una enfermedad hereditaria?
Sabemos que sí, recuerda que la hemoglobina es una proteína y las proteínas se generan a partir de la información codificada en nuestro ADN que se encuentra contenida en los cromosomas.
El grupo α, se localiza en el brazo corto del cromosoma 16 y el grupo β se localiza en el brazo corto del cromosoma 11.
Cuando hay una alteración en el material genético que codifica para las proteínas que constituyen la hemoglobina, ocurre una (mutación) que da como resultado a una proteína anormal.
Las mutaciones también pueden dar lugar a deleciones, que es la eliminación de la información genética para sintetizar proteínas, produciendo un desequilibrio en la cantidad de cadenas de globina.
Recapitula los conocimientos citados ¿Qué tipos de hemoglobinas existen?
Las diversas cadenas de globina se designan con letras griegas que se utilizan para describir la hemoglobina en particular; la hemoglobina del recién nacido en su mayor parte es hemoglobina fetal (HbF) y la hemoglobina para un niño mayor de 1 año que es igual a la del adulto, está representada por la hemoglobina A adulta (Hb A), entonces existen estos tipos de hemoglobinas:
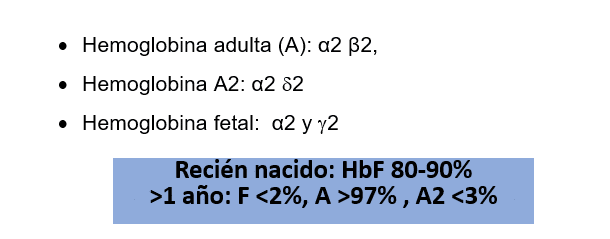
En los pacientes talasémicos, particularmente los Betalásemicos al estar disminuidas las cadenas betas, las cadenas alfas no encuentran con quien aparearse y se aumentan los otros tipos de hemoglobina especialmente la hemoglobina A2, que nos ayudará a diagnosticar este tipo de condiciones, por ello la importancia en que aprendas a describir los tipos de hemoglobinas.
Creo, que ya estamos preparados para hablar del diagnóstico: ¿Sabes Cómo se hace el diagnóstico de las hemoglobinopatías?
La talasemia es una anemia hemolítica intracorpuscular extravascular; se llama intracorpuscular porque el defecto se halla en el citoplasma del glóbulo rojo, que es igual a decir que se halla en el corpúsculo y es extravascular porque dada la vida media menor de 120 días del eritrocito talasémico, su destrucción como glóbulo rojo anormal lo hace el cuerpo en el bazo y no en el vaso sanguíneo.
Los pacientes con hemólisis cursan con palidez, ictericia y esplenomegalia,
Los casos severos de talasemia desarrollarán alteraciones faciales producto de la hematopoyesis extramedular que es la que se da fuera de los huesos largos, debido al agotamiento en estos sitios, requiriendo de apoyo de otras estructuras que contribuyeron en vida intraembrionaria en el proceso hematopoyéico, como el hígado, el bazo y los huesos planos, de allí la fascie de estos pacientes.
Existen otros tipos de complicaciones derivadas de la enfermedad y de la terapia transfusional o trasplante siendo esta última terapia la opción curativa definitiva.
Piensa ¿Cuál será la prueba en la que podemos analizar los diferentes tipos de hemoglobinas?
La electroforesis es la prueba más relevante para el diagnóstico de las hemoglobinopatías.
Existen varias técnicas para identificar la cantidad de hemoglobina A2, hemoglobinas anormales o inestables, entre ellas podemos citar las que tienen en cuenta el pH por eso se habla de electroforesis en medio alcalino y ácido las cuales pueden detallar los subtipos de hemoglobina con base a su carga de aminoácidos.
La hemoglobina es una molécula cargada negativamente (-) y en un campo eléctrico migra hacia la carga positiva (+); las variantes de la hemoglobina A con distintas cargas en su superficie se separan de la hemoglobina A y, por tanto, todas las detectadas son diferentes a ellas. Puede haber hemoglobinas anormales sin cambio en la carga, y por esto no serán diferenciadas y requerirán un buffer distinto dado en medio ácido para poder discriminar las HbC, S, A y F, y también los otros tipos de hemoglobinas que corren en el mismo sitio, como, por ejemplo: con la que la Hb A corren igualmente las Hb D, E, G, Leopore, H y J; la hemoglobina Bart´s se sitúa en la misma posición de la hemoglobina F. Otras técnicas utilizadas son la electroforesis capilar y la cromatografía líquida de alta resolución, esta última permite un análisis menos subjetivo que los métodos electroforéticos aislando mejor las variantes de hemoglobinas.
Disponiendo de todo este soporte, sé que ya cuentas con varias posibilidades diagnósticas, para estudiar a un paciente con microcitosis e hipocromía ¿Recuerdas cuáles son?
El diagnóstico diferencial de microcitosis e hipocromía en los niños incluyen varios diagnósticos como:
- Ferropenia
- Talasemia
- Intoxicación por plomo
- Anemia sideroblástica
- Otros
Es indispensable confirmar si hay una ferropenia para no ofrecer hierro innecesario a los pacientes talasémicos, como también poder administrar hierro a los que sí lo requieren.
Debemos incluir dentro de los diagnósticos diferenciales de los pacientes con microcitosis e hipocromía, quienes además tienen ferritina normal, a la talasemia.
Sabemos que estás listo para hacer un diagnóstico oportuno y certero. Acuérdate de disponer en tu diagnóstico las siguientes herramientas:
- Historia clínica completa investigando la ascendencia de los padres y sus antecedentes.
- Examen físico exhaustivo buscando la triada:
a. Anemia
b. Hemólisis
c. Esplenomegalia - Hemograma donde puedas verificar la presencia de la anemia, hipocromía y microcitosis.
- Extendido de sangre periférica que te describirá la hipocromía, microcitosis, poiquilocitosis (células en diana o target características de la talasemia).
- Electroforesis de hemoglobina que te ayudará a cuantificar la HbA2 (una HbA2 normal o bordeline no excluye el diagnóstico).
- Estudios moleculares en casos específicos.
Conclusiones
La talasemia es una enfermedad mundial, su alta frecuencia es un reflejo de la selección natural combinada de un número alto de matrimonios consanguíneos, productos de la migración en muchos países, Colombia es uno de ellos.
Para el clínico, la Talasemia requiere un reto en el diagnóstico diferencial entre anemia ferropénica, con el fin de establecer una terapéutica adecuada y seguimiento de una enfermedad crónica que tiene una presentación clínica variada, desde lo asintomático, hasta mostrarse simplemente con hallazgos de microcitosis e hipocromía en un hemograma o debutar con la condición severa que requiere terapia múltiple transfusional con hemocomponentes y trasplante.
El manejo integral depende de un grupo multidisciplinar, donde participamos todos los entornos, en la detección temprana, además del área de la salud, con estrategias tales como tamizaje neonatal de hemoglobinopatías, la cual está disponible en muchos países donde la Talasemia es endémica, pero debe ser fortalecida en Colombia.
Estamos convencidos que la detección precoz de niños con hemoglobinopatías severas cuya supervivencia podría llegar a ser menor de 5 años cuando no hay opción curativa de tratamiento, servirá para disminuir o anular importantes desenlaces.
Acuérdese que usted hace parte del proceso !!!
Agradecimientos
Extendemos nuestra gratitud, al Hospital Militar Central (HOMIL), Departamento de Pediatría Especialidades y Supraespecialidades, Servicio de Hematooncología Pediátrica, Departamento de Patología, Laboratorio Clínico e Inmunología, Servicio de Enfermería, Estudiantes, Grupo de Comunicación y de Educación Continuada; así mismo a nuestro cuerpo de Conferencistas y al Colegio Nacional de Bacteriología (CNB) por hacer posible, llevar información útil sobre Talasemia a todos en Colombia y países hermanos.